Synthesis of Molecularly Imprinted Polymer for Selective Solid-Phase Extraction of Salbutamol from Urine Samples
ALI MOHAMMADI*†, TAHER ALIZADEH‡, RASSOUL DINARVAND†,
MOHAMMAD REZA GANJALI‡ and RODERICK B. WALKER§
Department of Drug and Food Control, School of PharmacyTehran University of Medical Sciences, P.O. Box: 14155-6451, Tehran 14174, IranFax: (98)(21)6461178; Tel: (98)(21)6959090; E-mail: alimohammadi@tums.ac.ir
A highly selective methacrylic based molecularly imprinted polymer
(MIP) was synthesized and applied for the separation and the pre-concen-tration of salbutamol in urine samples. Spectrophotometric determinationof salbutamol was achieved using 2,6-dichloroquinone chlorimide ascolorimetric reagent. The detection limit of the method was ca. 13 ngmL-1 in urine after pre-concentration of the samples by MIP-SPE andanalysis with an optimized and sensitive spectrophotometric method. The linear dynamic range for salbutamol determination in urine was0.04-0.75 µg mL-1. The recovery for the affinity based solid-phaseextraction (SPE) with the MIP was more than 96 %. Key Words: Salbutamol, 2,6-Dichloroquinone chlorimide, Molecularly imprinted polymer, Solid-phase extraction. INTRODUCTION
Salbutamol [2-(tert-butylamino)-1-(4-hydroxy-3-(hydroxyl ethyl)phenyl)ethanol],
also known as albuterol, is a β-adrenergic receptor agonist, which is used as abronchodilatator in the treatment of reversible bronchospasm1. The most commonlyused methods for the analysis of salbutamol are gas and liquid chromatographicmethods, flow injection analysis, chemiluminescence, electrophoresis, thin layerchromatography, normal and derivative spectrophotometric methods. Furthermorethe use of spectrophotometric detection for salbutamol based on the formation ofcolour complexes has also been reported2-7. The methods have been reported arenon-selective and are affected by the presence of compounds that contain redoxreactive functional groups.
†Nanotechnology Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences,
P.O. Box 14155-6451, Tehran 14174, Iran.
‡Center of Excellence in Electrochemistry, Faculty of Chemistry, University of Tehran, Tehran,
§Department of Pharmaceutics, Faculty of Pharmacy, Rhodes University, Grahamstown, South
Molecular imprinting has been recognized as a promising technique for the
development of molecular recognition-based separations and sensing systems, wherethe molecule to be recognized is added to a reaction mixture consisting of a cross-linking agent, a solvent and a functional monomer that possesses functional groupscapable of interacting with the target molecule. Binding sites in the resultant polymerinclude functional groups that originate from the functional monomer and whichcan be constructed according to the shape and chemical properties of the targetmolecule. Following removal of the target molecule the molecularly imprintedcomplementary binding sites exhibit a high degree of selectivity and affinity for thetemplate molecule8. A high degree of selectivity is particularly desirable whenextraction of a single compound from a complex matrix is required. Consequentlythe use of molecularly imprinted polymers (MIPs) in such analyses is an attractiveoption, particularly when used as molecularly selective sorbents in solid-phaseextraction (SPE)9. The aim of the present research was to study the affinity-basedseparation and pre-concentration of salbutamol in urine samples using MIP-SPEwith spectrophotometric detection based on the reaction of 2,6-dichloroquinonechlorimide (DCQ) (an electron acceptor) with salbutamol10 (an electron donor). EXPERIMENTAL
A Shimadzu Model 160A UV-Visible double beam spectrophotometer with
quartz cells of 1.0 cm was used for the analysis of all samples. A pH meter wasused for the pH checking of the universal and the ammonia buffer solutions. Digitalscales for precise weighting, soxhlet apparatus, laboratory mortar, laboratory pestleand water bath for the MIP synthesis were also used.
All chemicals and solvents used were at least of analytical or pharmaceutical
grade. All solutions were prepared in double distilled water. 2,6-Dichloroquinonechlorimide (DCQ) was supplied by the Aldrich Company (Natick, MA, USA). Freshsolutions of DCQ (0.1 % w/v) in isopropanol, universal and ammonia buffer solutionsof pH 9.0 were freshly prepared immediately prior to use. Standard solution ofsalbutamol (2.0 mg mL-1) was prepared in water. All solvents and the methacrylicacid were purchased from the Merck Company (Darmstadt, Germany). Ethyleneglycol dimethacrylate and azo-N,N'-diisobutyronitrile (AIBN) were obtained fromthe Fluka (Buchs, Switzerland). Molecularly imprinted polymer (MIP) preparation: To manufacture the
salbutamol imprinted polymer AIBN (initiator 0.1 mg) and 0.5 mmol of salbutamolwere dissolved in 5 mL of chloroform in a 20 mL glass tube. Methacrylic acid(MAA, 1.7 mmol) and ethylene glycolmethacrylate (EDMA, 10.5 mmol) were addedto this solution and the mixture was degassed using nitrogen sparging for 10 min. The tube was sealed and heated in water bath at 50 ºC for 30 h. At the conclusion ofthe heating process a dense rigid polymer network that was white in colour hadbeen produced. The tube was destroyed and the monolithic polymer that remainedwas ground using a mortar and pestle. In order to remove the residual monomers
Solid-Phase Extraction of Salbutamol from Urine Samples 2877
and salbutamol the polymer was subjected to extraction using a Soxhlet extractionapparatus and a mixture of methanol-acetic acid in an 8:1 v/v ratio. In order toascertain whether the extraction process was effective the concentration ofsalbutamol in the extraction solvents was determined using colorimetric methodbased on the charge transfer complex formation that occurred between salbutamoland the DCQ reagent. The polymer was ground once again using a mortar andpestle to effect particle size reduction and the resultant material was sieved througha 40 µm sieve with the aid of distilled water. The resultant fine particles wereremoved by decantation in acetonitrile and used to evaluate their potential forextraction of urine samples. A control, non imprinted polymer (NIP) was also manu-factured in a similar manner by polymerization without salbutamol and the controlpolymer was used to establish the existence of nonspecific binding sites forsalbutamol. RESULTS AND DISCUSSION Spectrophotometric determination of salbutamol: A previously reported
procedure10 was used as the method of choice for the determination of salbutamolin the presence of the DCQ reagent. Since DCQ is an electron acceptor and thebenzene ring in salbutamol and isoxsuprine molecules consist of the electron richgroups, a π→π* CT complex can be formed. The chemical structure of salbutamoland isoxsuprine in addition to that of the charge transfer complex that is formedbetween salbutamol and DCQ are depicted in Fig. 1.
Chemical structure of isoxsuprine and salbutamol molecules and the chargetransfer complex formation reaction between the DCQ and the salbutamol
MIP evaluation using colorimetry: The molecular recognition ability of the
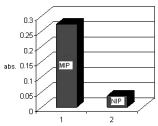
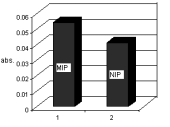
MIP was evaluated using colorimetry. A fixed amount of MIP powder was added toa fixed volume of aqueous sample, containing 2 µg mL-1 of salbutamol. After 1 h,the target molecule had been adsorbed was removed from the MIP using methanol. This procedure was also performed with the NIP. The results of this evaluation aredepicted in Fig. 2 and reveal that the salbutamol reaction with the DCQ followingextraction from an aqueous solution with MIP and that of the NIP, respectively. It isclearly evident that the capability of the MIP to adsorb salbutamol is substantiallyhigher than that of the NIP thereby providing evidence for the existence of a selectivesite for salbutamol sequestration on the MIP as compared to that of the NIP. Inanother evaluation of the selectivity of MIP for salbutamol the adsorption ofisoxsuprine, as structurally similar molecule to salbutamol was investigated. Ascan be seen in Fig. 3 there is minimal difference for the adsorption of isoxsuprinebetween the MIP and NIP materials used. Therefore the selective sites for salbutamolsequestration that are present on the MIP, do not appear to demonstrate as high anaffinity for the uptake of isoxsuprine as for salbutamol. Furthermore, it can bededuced that mechanism of adsorption of isoxsuprine are similar for both the MIPand NIP polymeric materials.
Comparison of salbutamol adsorption Fig. 3. Comparison of isoxsuprine adsorptionusing the MIP and NIP polymers. The
Calibration curve and detection limit using the MIP-SPE: The calibration
for the MIP-SPE determination of salbutamol was carried out under the specifiedoptimal conditions. The calibration graph is characterized by a linear range from0.04 to 0.75 µg mL-1 and an equation described by A = 0.734 C (µg mL-1) + 0.111with correlation coefficient equal to 0.9988. The detection limit of this method wascalculated about 13 ng mL-1.
Solid-Phase Extraction of Salbutamol from Urine Samples 2879
Interference studies: All analyses, in which the reactivity of the salbutamol
phenolic group is essential to generate a response, will be negatively affected bythe presence of other compounds that are readily oxidized. Therefore the use of theMIP-SPE technique provides an opportunity to overcome this challenge assalbutamol is preferentially adsorbed whereas other similar materials will not be. The use of the proposed MIP-SPE method was applied to the determination ofsalbutamol in urine samples without any additional analytical artifacts or challenges. In addition, the presence of compounds such as starch, lactose, glucose, ascorbicacid, urea and uric acid in concentrations up to 100 times higher than that ofsalbutamol did not result in any interference in analysis of urine using this method. Analytical application: The proposed method has been successfully used for
the determination of salbutamol in spiked human urine samples. In order to evaluatethe potential of this method, an aliquot of aqueous salbutamol standard solutionwas added to urine and diluted to between 5 to 20 times to reach a total samplevolume of 150 mL and a salbutamol concentration range of between 0.04-0.75 µgmL-1. 0.1 g of MIP was added to the urine and the container was agitated for 0.5 h. The sample was filtered through filter paper and washed with 4 mL ofdichloromethane and then recovered with 3 mL methanol. Following extraction thesample was transferred to a 5 mL volumetric flask and evaporation of the methanolachieved using nitrogen 1 mL of buffer, prepared as previously described and 1 mLof the DCQ reagent solutions were added and to achieve a total volume to 5 mL. The colour was allowed to develop over 15 min and the resultant absorbance wasmeasured at a wavelength of 620 nm. A calibration curve was constructed and thesalbutamol concentration was calculated. Table-1 lists a summary of the salbutamolconcentrations in spiked urine samples.
CONCENTRATION OF SALBUTAMOL IN SPIKED URINE
Conclusion
In the present study a molecular imprinted polymer was prepared and used for the
determination of salbutamol in human urine. The evaluation of the MIP/salbutamolreaction by colorimetric determination was shown to have a degree of selectivity ofdue to the affinity of the novel sorbent for salbutamol. It has been proven that bycombining affinity based MIP-SPE and a simple spectrophotometric method a precise,selective and sensitive determination of salbutamol in low concentration is possible. ACKNOWLEDGEMENT
The authors would like to acknowledge the financial assistance from the Medical
Sciences University of Tehran, Tehran, Iran. REFERENCES
R. Berges, J. Segura, X. De La Torre and R. Ventura, J. Chromatogr. B: Biomed. Appl., 723, 173 (1999).
K.K. Bhatt, S.A. Shah and S.S. Pandya, Indian Drugs, 36, 524 (1999).
K.P.R. Chowdary and R.G. Devala, Indian Drugs, 34, 283 (1997).
R.S. Bakry, O.A. Razak, A.F.M. El Walily and S.F. Belal, J. Pharm. Biomed. Anal., 14, 357 (1996).
T.D. Burns, N. Chimpalee, D. Chimpalee and K. Leiwongcharoen, Anal. Chim. Acta, 260, 65 (1992).
N.V. Naidu, D.V. Naidu, C.V. Rajeswari and P.R. Naidu, Acta Chim. Hung., 126, 821 (1989).
N. Geeta and T.R. Baggi, Microchem. J., 39, 137 (1989).
T. Takeuchi and J. Haginaka, J. Chromatogr. B, 728, 1 (1999).
A. Beltran, E. Caro, R.M. Marcé, P.A.G. Cormack, D.C. Sherrington and F. Borrull, Anal. Chim. Acta, 597, 6 (2007).
10. G.G. Mohamed, S.M. Khalil, M.A. Zayed and M. Abd El-Hamid El-Shall, J. Pharm. Biomed.Anal., 28, 1127 (2002).
(Received: 17 May 2008; Accepted: 19 January 2009)AJC-7134
Using Terahertz Pulse Spectroscopy to Study the CrystallineStructure of a Drug: A Case Study of the Polymorphs ofRanitidine HydrochlorideP.F. TADAY, I.V. BRADLEY, D.D. ARNONE, M. PEPPERTeraView Limited, 302/304 Cambridge Science Park, Milton Road, Cambridge, CB4 0WG, UKReceived 22 July 2002; revised 23 October 2002; accepted 22 November 2002ABSTRACT: We describe the application of Terahertz pu
 MIP evaluation using colorimetry: The molecular recognition ability of the
MIP evaluation using colorimetry: The molecular recognition ability of the